Intro to RNA-Seq¶
learning-objectives
- Know what the transcriptome is
- Understand how changes in the transcriptome reflect a cell’s condition (e.g. developmental state, disease state, etc.)
- Know how an RNA-Seq experiment can reveal information about the transciptome
RNA-Seq - A look into the transcriptome¶
DNA makes RNA makes Protein. This “Central Dogma” of molecular biology is a fundamental concept in molecular biology. With the development of DNA sequence, we can explore the genome - all of the DNA in an organism. However, DNA is only the starting point. To understand what genes are active, and under what circumstances, we have to know what genes are being transcribed into messenger RNA. Starting from the first cell - in for example a human or a mouse - every other cell differentiates itself not by having different DNA, but by expressing (transcribing) those genes differently. A cell in the liver has the same DNA instructions as a neuron in the brain. However the genes being expressed differ greatly between these cells. The sum of all RNAs being expressed in a cell is known as the transcriptome.
Gene transcription also changes as the cell responds to its environment. As cells are exposed to hormones for example a mammal undergoing the transition to reproductive maturity, cells may change their pattern of gene expression. Hormones are not the only things that will change gene expression. Nearly any environmental stimulus (e.g. presence of a nutrient like glucose, pH changes that correspond to the level of oxygen) might result in a change in expression.
Besides developmentally or environmentally induced changes in gene expression, Disease is another condition which can be marked by characteristic changes in gene expression. Cells exposed to viruses or bacteria may express genes associated with resistance and/or stress. A cancerous cell may be expressing too much (overexpression) of a gene, or may not be expressing other genes.
RNA-Seq Experiment¶
RNA-Seq is a widely-used experiment that has become one of the most important ways to characterize the transcriptome. There are several variations, details, and statistics we are glossing over, but at it’s most basic we use an RNA-Seq experiment to ask the question - “does the gene expression in a control sample differ from the gene expression in an experimental sample?”
Note
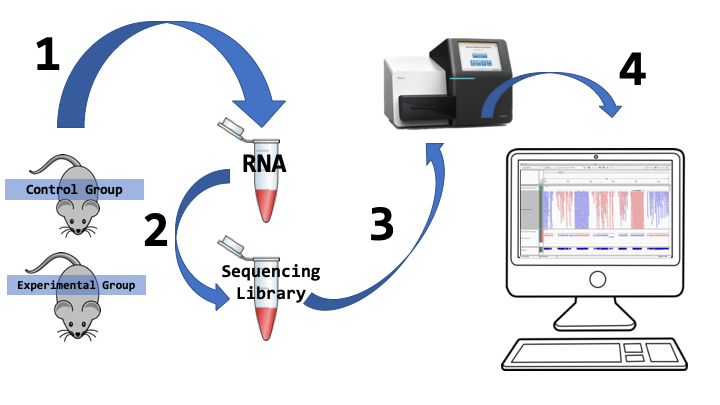
RNA-Seq Experiment - Basic Steps
At its simplest RNA-Seq involves 4 major steps:
- RNA is extracted from an organism of interest. Choices must be made at this stage about sampling of the organisms, since not all RNAs of interest may be present in every tissue and at all times. RNA is also easily degraded, and the extraction procedure can introduce biases into the collected sample. How we extract samples defines our experimental design. Typically we will sample at least two distinct groups (e.g. wild-type/mutant, healthy/disease, or control/experimental). We also will have several biological replicates since all our sampling will contain biases.
- Several biochemical steps are taken to prepare extracted RNA into a sequencing library. RNA is converted into stable cDNA and undesired RNA (e.g. ribosomal RNA) is removed. Adaptors are attached to the cDNAs.
- There are a variety of sequencing platforms that can generate the sequence of a prepared library. In the popular illumina protocol, millions of short cDNA fragments will be generated (usually 100-300bp in size).
- Following sequencing, the RNA-Seq reads are quality controlled and then software is used to align and or assemble these reads for further analysis.
Recommended reading: RNA-Seq: a revolutionary tool for transcriptomics Wang Z. et.al. Nat.Rev.Genetics 2009;(10):57-63. https://www.nature.com/articles/nrg2484
Example Experiment: Leptin expression in mouse tumor¶
Sample data
Mouse tumor sample dataset
In this lesson, we will use an RNA-Seq dataset collected from tissue sample in mice on a high-fat diet. In the paper that describes this dataset investigators were looking to understand how human obesity plays a role in colon cancer. Two genes including leptin and Wnt were found to be elevated in mice colon tumor samples when the mice were on a high-fat diet. These findings are important because in human colon cancers, increased expression of leptin is associated with a lower survival rate.
Sample data citation: Penrose HM, Heller S, Cable C, Nakhoul H, Baddoo M, Flemington E, Crawford SE, Savkovic SD. High-fat diet induced leptin and Wnt expression: RNA-sequencing and pathway analysis of mouse colonic tissue and tumors. Carcinogenesis. 2017 Mar 1;38(3):302-311. doi: 10.1093/carcin/bgx001. PMID: 28426873; PMCID: PMC5862315. [Leptin paper]
Using This Lesson¶
In this lesson, we will be looking at gene expression using RNA-Seq data from mouse tumor tissue. There is sequence from six samples (3 tumor samples, 3 non-tumor samples) from the mouse leptin experiment. To analyze the data we will use Jupyter notebooks to run several bioinformatics tools:
Notebook Tools used Goal Notebook-0 Import file from NCBI Sequence Read Archive NCBI SRA tools The Sequence Read Archive (SRA) is a massive repository of DNA and RNA sequence data. We will show you how data from SRA can be imported and analyzed. Notebook-1 Running FastQC FastQC We will check the quality of our sequence data Notebook-2 Trimming Data Trimmomatic We will remove low-quality data from our experiment Notebook-3 RNA-Seq from scratch - Kallisto Kallisto We will match RNA-Seq sequence from our experiment to the known mouse transcriptome Notebook-4 Visualize BAM file alignment at the Leptin gene locus genomeview, UCSC, IGV We will visualize how reads from our dataset map to the mouse genome at the leptin gene locus and see the difference in expression levels between samples Notebook-5 Backup Data iCommands This notebook is a backup allow you to import worked solutions/files for any analysis you can’t complete
Working with Notebooks¶
You will work through notebooks 1-4 in this lesson. Each notebook has instructions to follow as well as a companion page on this site with additional questions and background information.
Working as a team
Since we are working with a real dataset, some of the steps in this analysis can take several minutes (as long as 20 minutes per-sample for Notebook 3 Kallisto alignment). To save time, it may be best for each student to take one sample through all of the notebook steps.
Working as an individual
An individual can also take all 6 samples through all the notebook steps. Allow for extra time if you choose this strategy.
Worked results
Follow the instructions in notebook 5 to import the outputs from any notebook you did not complete. You can skip ahead or import files if something fails to work.
Questions¶
Question
- What is the transcriptome?
- What are some things that can alter a cell’s transcriptome
- What can an RNA-Seq experiment tell you?
- What are the major steps in an RNA-Seq experiment?
Bonus
In an RNA-Seq experiment we may sample a tissue (e.g. a piece of colon tissue, a biopsy of tumor tissue, etc.) How would sampling a tissue (made up of many cells) be different from measuring the trancriptome of an individual cell?